Interval joins
Overview
Consider datasets s1 and s2, storing genomic intervals, such as |s1|<|s2|. Let’s assume that their structure contains necessary genomic coordinates (chromosome, start position, end position) along with optional additional interval annotations (e.g. targetId).

Tables structure
Dataset `s1` and `s2`. Both storing genomic intervals with necessary genomic coordinates and optional annotations
Our goal is to efficiently execute a query as shown below:
SELECT s2.targetId,count(*)
FROM reads s1 JOIN targets s2
ON s1.chr = s2.chr
AND s1.end >= s2.start
AND s1.start <= s2.end
GROUP BY targetId;
Algorithm
At it’s core SeQuiLa’s range joins are based on IntervalTree data structure. The main idea of the algorithm is to transform dataset s1 into a broadcastable structure of an interval forest (a hash map of interval trees, each representing one chromosome). The intervals from dataset s2 can be efficiently intersected with the constructed interval forest.
An interval tree is a tree data structure to hold intervals. It is a augmented, balanced red-black tree with low endpoint as node key and additional max value of any endpoint stored in subtree.
Each node contains following fields: parent, left subtree, right subtree, color, low endpoint, high endpoint and max endpoint of subtree.
It can be proved that this structure allows for correct interval insertion, deletion and search in :math:O(lg n) time ([CLR]_)
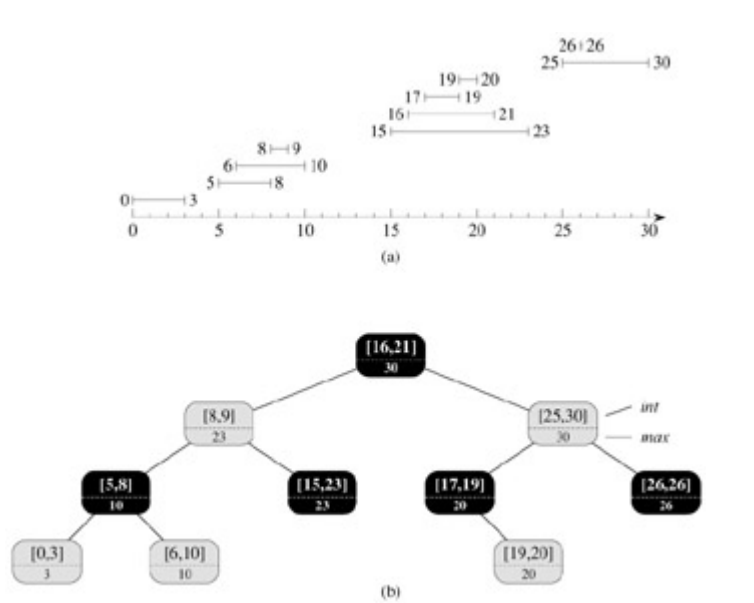
Interval tree
An interval tree. On the top: A set of 10 intervals, shown sorted bottom to top by left endpoint. On the bottom the interval tree that represents them. An inorder tree walk of the tree lists the nodes in sorted order by left endpoint.
Our implementation of IntervalTree is based on explanations in [CLR]_ although it is extended in the following ways:
- data structure allows storing non-unique intervals
- data structure allows storing in the tree nodes additional interval annotations if they fit into dedicated Spark driver’s memory fraction
Let’s presume that we have a cluster with a Spark driver, three worker nodes and that the tables are partitioned between worker nodes. When interval query is performed, all dataset’s :math:s1 partitions are sent to the driver node on which interval forest is constructed (for each chromosome a separate interval tree is created). The forest is subsequently sent back to worker nodes on which efficient interval operations based on interval trees are performed. Depending on the strategy chosen by rule-based optimizer different set of columns are stored in tree nodes
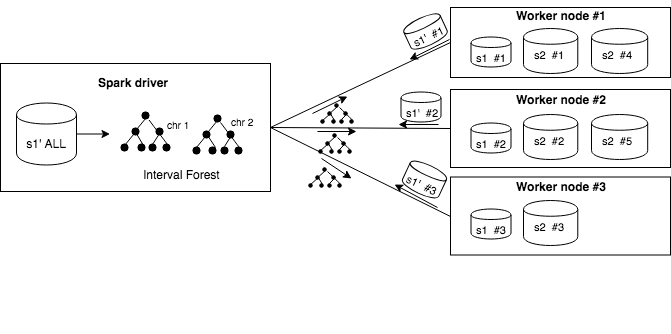
Broadcasting mechanism
Broadcasting interval forest to worker nodes.
.. [CLR] Cormen, Thomas H.; Leiserson, Charles E., Rivest, Ronald L. (1990). Introduction to Algorithms (1st ed.). MIT Press and McGraw-Hill. ISBN 0-262-03141-8
Optimizations
SeQuiLa package introduces a new rule based optimizer (RBO) that chooses most efficient join strategy based on
input data statistics computed in the runtime. The first step of the algorithm is to obtain value of maxBroadcastSize parameter. It can set explicitly by the end user or computed as a fraction of the Apache Spark Driver memory.
In the next step table row counts are computed and based on that table with the fewer rows is selected for constructing interval forest. This is the default approach - it can be overridden by setting
spark.biodatageeks.rangejoin.useJoinOrder to true. In this scenario no row counts are computed and the right join table is used for creating interval forest. Such an strategy can be useful in situation when it is known upfront which table should be used for creating a broadcast structure.
The final step of the optimization procedure is to estimate the row size and the size of the whole projected table.
If it fits into dedicated Spark Driver’s memory (controlled by maxBroadcastSize parameter) the interval forest is augmented with all columns from s1 (SeQuiLa_it_all strategy) completing map-side join procedure in one stage. Otherwise an interval tree is used as an index for additional lookup step before the equi-shuffle-join operation between s1 and s2 (SeQuiLa_it_int strategy).
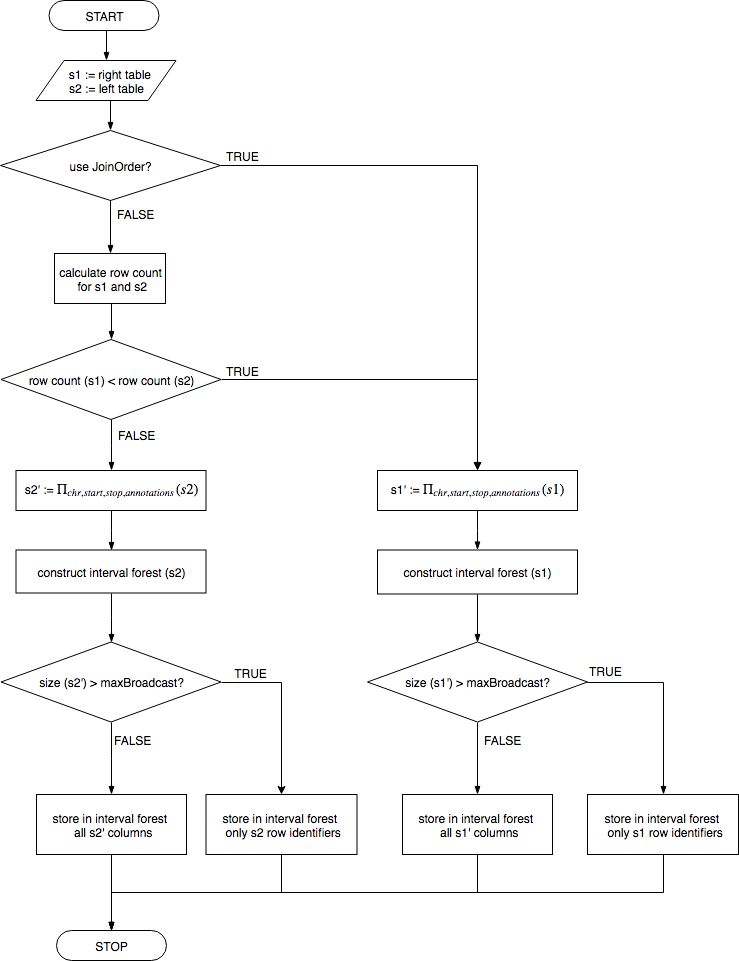
Optimization algorithm
Rule-based optimizer’s algorithm chooses the most efficient join strategy.
Custom interval structure
By default SeQuiLa uses Red-Black tree with intervals implemented in org.biodatageeks.sequila.rangejoins.methods.IntervalTree.IntervalTreeRedBlack<V>
class. However, it is possible to provide a custom interval structure and set it using spark.biodatageeks.rangejoin.intervalHolderClass
parameter. There are 2 prerequisites:
-
custom class must implement interface provided by the trait:
org.biodatageeks.sequila.rangejoins.methods.base.BaseIntervalHolder[V] -
node class used for storing intervals must extend
org.biodatageeks.sequila.rangejoins.methods.base.BaseNode[V]
Both Scala and Java classes are supported. Please use default interval tree implementation for reference.
Feedback
Was this page helpful?
Glad to hear it! Please tell us how we can improve.
Sorry to hear that. Please tell us how we can improve.